

Projects
Nucleosome positioning regulates genome functions
A major part of eukaryotic nuclear DNA is organized into nucleosomes that are not distributed randomly, but occupy defined positions with respect to the DNA sequence (for our review see Lieleg et al., 2015, Chromosoma). Such nucleosome positioning is fundamental to all genomic processes as it regulates the accessibility of DNA. For example, a transcription factor binding site may reside in a nucleosome free region and be readily bound by its corresponding factor in one cell but may be incorporated in a nucleosome and therefore be inaccessible in a different cell (Fig. 1).
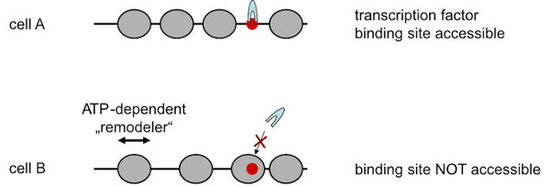
Figure 1. Positioning of nucleosomes (grey spheres) along DNA regulates DNA accessibility, e.g., to the binding site (red dot) of a transcription factor (grey arch). Nucleosome organization is remodeled by ATP-depenent nucleosome remodeling enzymes (“remodelers”).
We wish to understand which factors determine nucleosome positioning, especially how DNA sequence is read out and translated into nucleosome positioning, and how positioned nucleosomes in one biological state become remodeled into an altered organization in a different state. This is relevant, for example, in promoter regions where nucleosome organization primes or represses transcriptional activity as pioneered for the yeast PHO promoters (see below). Active eukaryotic genes show a stereotypical nucleosome positioning pattern with a nucleosome free region (NFR) just upstream of the transcription start site (TSS) and an array of regularly spaced nucleosomes over the gene (Fig. 2).
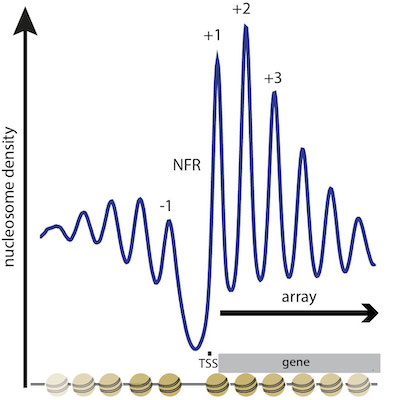
Figure 2. Stereotypical NFR-array nuleosome positioning pattern at the 5’ end of active genes. Composite plot of MNase-seq signal (nucleosome density) aligned at transcriptional start sites (TSS) and integrated over all genes in S. cerevisiae. Peaks correspond to positioned nucleosomes and are numbered (-1, +1, +2, ...) relative to the TSS. Troughs correspond to linker regions between nucleosomes or more extended nucleosome free regions (NFR), especially over active promoters. (modified from Lieleg et al., 2015, Chromosoma)
ATP dependent chromatin remodeling enzymes mediate nucleosome positioning and dynamics
Central players in nucleosome positioning and remodeling are ATP-dependent chromatin remodeling enzymes (“remodelers”, Fig. 3) that confer nucleosome sliding, regular spacing, (dis)assembly and histone exchange. We study the role of remodelers in nucleosome positioning since long (Korber et al., 2004, J. Biol. Chem.; Hertel et al., 2005, Mol. Cell. Biol.; Lantermann et al., 2010, Nat. Struct. Mol. Biol.; Wippo et al., 2011; EMBO J.; Zhang et al., 2011, Science; Pointner et al., 2012, EMBO J.; Lieleg et al., 2015, Mol. Cell. Biol.; Krietenstein et al., 2016, Cell; Knoll et al., 2018, Nat. Struct. Mol. Biol.; Oberbeckmann, Krietenstein et al., 2021, Nat. Comm., Oberbeckmann, Niebauer et al., 2021, Nat. Comm.) and identified that some remodelers, like the INO80 complex, are able to position nucleosomes in a sequence-guided way.
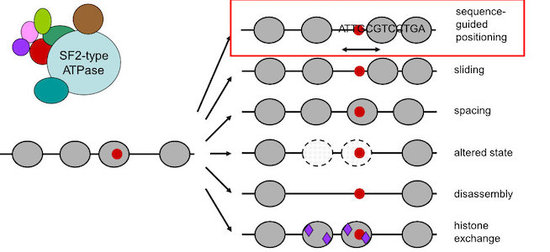
Figure 3. Nucleosome remodeling enzymes are often large complexes that contain SF2-type ATPases and remodel nucleosome organization in various ways. We identified (Krietenstein et al., 2016, Cell; Oberbeckmann, Krietenstein et al., 2021, Nat. Comm.) sequence-guided nucleosome positioning by remodelers (red box).
Remodeler mechanisms studied by pioneering genome-wide chromatin reconstitution and powerful yeast genetics
As model systems we use the unicellular yeasts Saccharomyces cerevisiae and Schizosaccharomyces pombe and combine their powerful genetic tools with biochemical techniques. Especially regarding the biochemical approach, we established a unique genome-wide in vitro reconstitution system that enables us to dissect the mechanisms of individual factors in nucleosome positioning and remodeling (Fig. 4 and Zhang et al., 2011, Science, Krietenstein et al., 2012, Meth. Enzymol., Krietenstein et al., 2016, Cell, Oberbeckmann et al., 2021, Nat. Comm.).
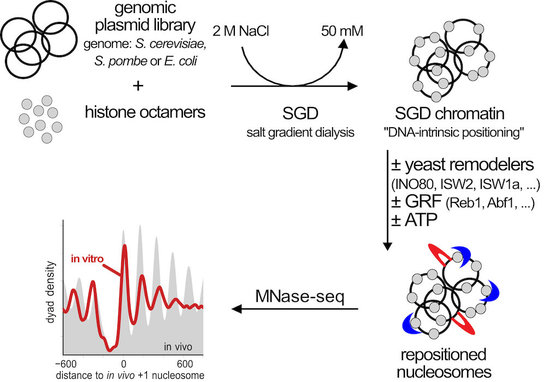
Figure 4. Genome-wide in vitro reconstitution approach to dissect the mechanisms of nucleosome positioning and remodeling. Genomic plasmid libraries are reconstituted with purified histone octamers into nucleosomes by salt gradient dialysis (SGD). The SGD chromatin is a proxy for nucleosome positioning by histone octamer binding preferences (“DNA-intrinsic positioning”) and serves as starting point for remodeling by a variety of purified factors that we add. Resulting nucleosome organization is analyzed by MNase-seq and other methods. (modified from Oberbeckmann, Niebauer et al., 2021)
With this genome-wide biochemistry approach we demonstrated (Fig. 5 and Krietenstein et al., 2016, Cell) that the stereotypical NFR-array nucleosome organisation can be generated by the interplay of remodelers, DNA sequence features and so called “barrier” factors (also known as general regulatory factors, GRFs).
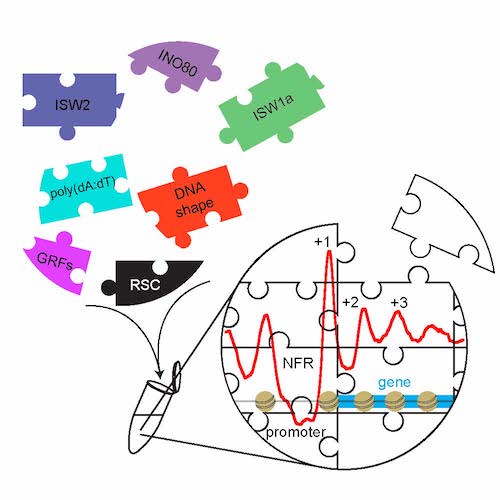
Figure 5. The combination of purified nucleosome remodeling enzymes (like ISW2, ISW1a, INO80, or RSC), general regulatory factors (GRFs = “barriers”), and DNA sequence features (like poly(dA:dT) elements or shape features) is sufficient to reconstitute in vitro (symbolized by Eppendorf tube) the basic nucleosome organization at active yeast promoters. Graphical abstract created by Nils Krietenstein and taken from Krietenstein et al., 2016, Cell.
Currently, we further used (Oberbeckmann, Krietenstein et al., 2021, Oberbeckmann, Niebauer et al., 2021) and expanded our in vitro reconstitution system to study the mechanisms of nucleosome positioning and remodeling. This approach is paralleled by in vivo mutational studies with yeast. We conceptualize remodelers as information processing hubs that read out various features of DNA and protein components in chromatin and position/remodel nucleosomes according to this information input (Fig. 6). This involves “ruler elements” in remodelers that enable to measure out and set distances between nucleosomes and other reference points, like barriers or neighboring nucleosomes.
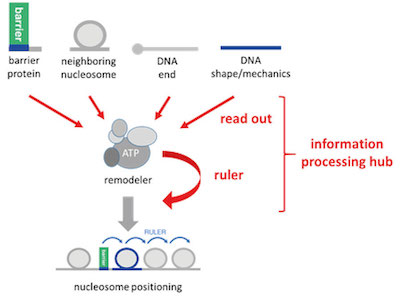
Figure 6. Remodelers conceptualized as information processing hubs. Remodelers read out molecular information from DNA or proteins, add their own built-in information (remodeler ruler) and process this combined information into nucleosome positioning or other remodeling outcomes. (modified from Oberbeckmann, Krietenstein et al., 2021)
Methods development
We continue to develop methods for monitoring nucleosome positioning and remodeling, for example, the first high resolution genome-wide mapping of nucleosome occupancy in absolute terms via DNA methyltransferase and restriction enzyme accessibility (Fig. 7 and Oberbeckmann et al., 2019). This also includes single molecule long read sequencing.
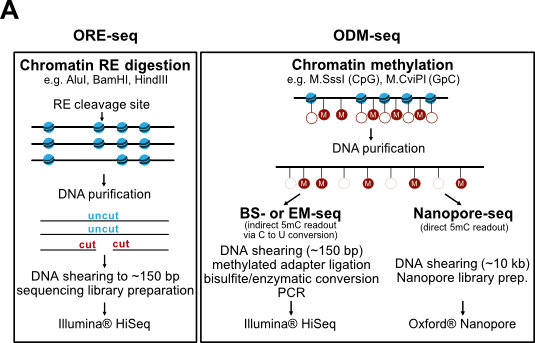
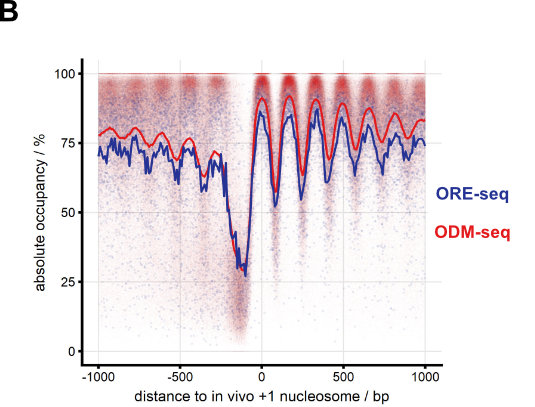
Figure 7. Absolute nucleosome occupancy map for S. cerevisiae. (A) Schematic of methods. ORE-seq and ODM-seq (occupancy measurement via restriction enzymes/methyl transferases and high throughput sequencing) monitor the ratio of accessible versus inaccessible DNA positions, which amounts to absolute occupancy. (B) Composite plots of ORE-seq/ODM-seq signal aligned at in vivo +1 nucleosome positions and integrated over all yeast genes (solid traces show average, individual dots show individual genomic sites). The pattern recapitulates classical MNase-seq patterns (Fig. 2), but the y-axis shows calibrated absolute occupancy for the first time. (modified from Oberbeckmann et al., 2019, Genome Res.)
Our former graduate student Corinna Lieleg initiated a collaboration with the group of Hendrik Dietz (Technical University of Munich). The Dietz group specializes in nano-structures based on complementary DNA strand hybridizations (“DNA origamis”) and explores their manifold applications. In collaboration with us, the Dietz group established a device that can be used as a molecular force spectrometer for analyzing nucleosome properties like the energy landscapes of stacking and unwrapping (Fig. 8 and Funke et al., 2016, Science Advances; Funke et al., 2016, ACS Nano Letters).

Figure 8. DNA origami force spectrometer allows direct measurement of nucleosome-nucleosome interactions (double headed arrow). Nucleosomes were attached to DNA origami via single strand hybridization handles. Red spiral symbolizes torsional spring of DNA origami device that counteracts nucleosome-nucleosome interactions. Red and green spheres show positions of dyes that allow monitoring of opening angles via FRET. Taken from Funke et al., 2016, Science Advances.
Yeast PHO promoters: classical paradigm for promoter regulation through nucleosome remodeling
Historically, our group originated in 2005 from the group of Wolfram Hörz, who pioneered the S. cerevisiae PHO5 and PHO8 promoters as classical model systems for the role of chromatin in gene regulation (for review see Korber and Barbaric, 2014, Nucl. Acids Res.). These promoters harbor positioned nucleosomes that control access to the transactivator Pho4 and to the transcriptional machinery. Upon promoter induction, these nucleosomes become remodeled, which leads to an extensive DNaseI hypersensitive site (Fig. 9).
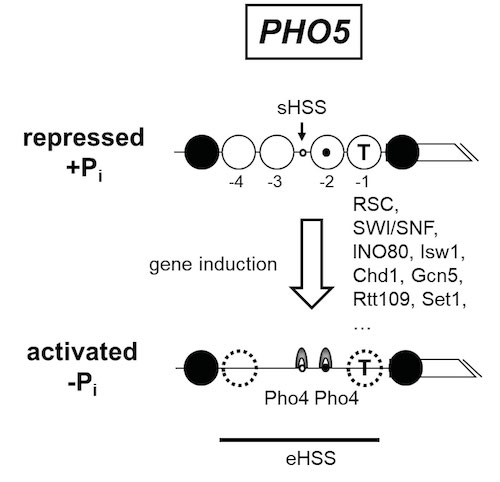
Figure 9. Schematics of the chromatin organization at the S. cerevisiae PHO5 promoter in its repressed and activated state. In the repressed state, the promoter is organized into positioned nucleosomes (open and closed large circles, numbered -1 to -4 relative to the gene (broken box)) with a short hypersensitive site (sHSS). Nucleosomes -1 and -2 occlude the TATA box (T) or one Pho4 binding site (black dot), respectively, while another Pho4 site (white dot) is constitutively accessible in the sHSS. The promoter is activated upon phosphate depletion of the cells, which activates the transactivator Pho4 (grey arches) and leads to complete eviction or partial remodeling (stippled circles) of the positioned nucleosomes . The resulting extensive hypersensitive site (eHSS, bold horizontal line) is a hallmark of the activated promoter. This chromatin transition involves a surprisingly complex cofactor network, e.g., the remodelers RSC, SWI/SNF, INO80, Isw1, Chd1, the histone acetyltransferases Gcn5 and Rtt109, the histone methyltransferase Set1 and probably even more.
The Hörz group elucidated many aspects of this remodeling, often in collaboration with the group of Slobodan Barbaric (University of Zagreb, Croatia). We continue this collaboration and the study of the mechanisms of PHO promoter remodeling by in vitro and in vivo techniques (Korber et al., 2004, J. Biol. Chem.; Hertel et al., 2005, Mol. Cell. Biol.; Korber et al., 2006, J. Biol. Chem.; Barbaric et al., 2007, J. Biol. Chem.; Wippo et al., 2009, Mol. Cell. Biol.; Ertel et al., 2010, Mol. Cell. Biol.; Musladin et al., 2015, Nucl. Acids Res.). Recently, we collaborated with the theoretical biophysics group of Ulrich Gerland (Technical University of Munich) in a modeling approach that allowed to assess the effective nucleosome configuration dynamics in the repressed and activated state, which are otherwise not tractable by experiments (Fig. 10). Surprisingly, this modeling suggests that PHO5 promoter opening is regulated by a change in nucleosome assembly, rather than disassembly, which challenges the classical view that remodeler recruitment by Pho4 upon induction increases nucleosome eviction.
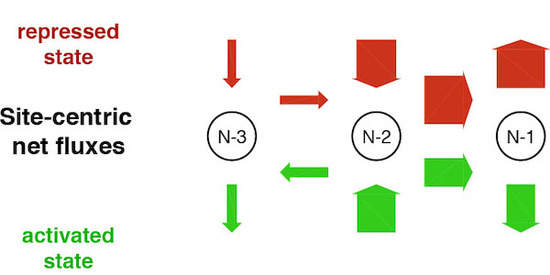
Figure 10. Site-centric net fluxes of nucleosomes -1 to -3 at the repressed or activated PHO5 promoter. Experimental data from three independent techniques and for several PHO5 promoter states were fit to an unbiased collection of models that comprise nucleosome assembly, disassembly and sliding (“regulated on-off-slide” models). Only 7 out of 68,145 models fit all data. Surprisingly, all of them contained a regulated assembly, rather than disassembly parameter (modified from Wolff et al., 2021, eLife).